Добрый день!
Моей второй дочери 1г2м. А началось все так.
Беременность 1 - роды со стимуляцией. 1 период(безводный) - 4 часа, 2 период(роды) - 10 мин. По Апгара - 8-9. (в 3 года у ребенка начались приступы, диагноз эпилепсия. На ЭЭГ - очаг. КТ - без патологии. Сейчас ребенку 7 лет, 3,5 года - ремиссия, очага на ЭЭГ нет). Ребенок развит. Закончили дет.сад, в этом году в школу.
Беременность 2 - мед.аборт
Беременность 3 - прерывание беременности по мед.показаниям в 24 нед. (УЗИ - ананцефалия)
Беременность 4 - Замершая 9 нед.
Беременность 5 - готовилась как в космонавты, все анализы отличные, токсикоза нет, работала до последнего дня. Роды со стимуляцией. 1 период(безводный) - 10 часов, 2 период(роды) - 25 мин. По Апгара - 7-8. Небольшое обвитие, задержка плечиков, выписали в срок. Дома все нормально, но с 3 месяцев стали ставить ЗПМР. В 5 мес - кортексин, в 7 - церебролезин. При этом несколько курсов массажа. к 10 мес. стали садиться, но у опоры не вставала, и вобще - какое-то безразличие, а может просто спокойствие. в 11 мес. начались приступы, поступили в больницу с судорожным синдромом. Результаты обследований:
НСГ: асимметрия боковых желудочков головного мозга.
ЭКГ: в пределах нормы.
ЭхоЭГ: смещение срединных структур головного мозга не выявлено.
ЭЭГ: грубые нарушения биоэлектрической активности головного мозга с признаками эпилептизации мозга. Паттерн ЭЭГ вспышка-подавления (вспышка представлена эпилептическими комплексами) на протяжении всей записи ЭЭГ.
МСКТ-ангиография: врожденный порок развития: сосудистая мальформация? опухолевидное образование?
Офтальмолог: глазное дно без патологии.
Назначение: депакин-хроно, пантогам, витамин Д3. Высокопольное МРТ.
В Новосибирске прошли МРТ с контрастом,
диагноз: Признаки узловой гетеротопии правой затылочной области дифференцировать с объемным образование не накапливающим контрастное вещество; рекомендован МРТ контроль в динамике. Внутренняя гидроцефалия.
Дома приступы пока сохранились, но бывают с разной периодичностью, то раза 3 в день, а то каждые 1.5-2 часа. и Интенсивность разная, но после Депакина они стали послабее, т.е. ее уже не так сильно "тянет" и по длительности 1-2 мин. А может и просто вздрогнуть и все. Приступы всегда были без потери сознания. Т.е. происходит запрокидывание головы назад и вправо, клонические подергивания мышц плечевого пояса, движение глаз влево-вниз. Приступ протекал в виде серий таких движений с периодичностью 1- 2 сек. по 5-6 раз. Все вместе занимало 1-2 мин. Но кроме приступов у нас еще проблема - зрение, т.е. она как будто не видит, хотя со стороны глаз патологий нет, и бывают моменты, когда кажется, что она смотрит. Нейрохирург сказал, что из-за врожденной патологии ГМ у нее выпадает часть поля зрения.
В настоящий момент только противосудоржная терапия, оформляем инвалидность, ждем консультацию эпилептолога (там очередь) и очередное ЭЭГ, чтоб посмотреть динамику.
А вопрос у меня такой: наши врачи говорят, что кроме противосудоржной терапии больше никакого лечения нет, прогнозы - только неблагоприятные, у нас в городе детей с такой патологией нет, и в инете почти нет инфы об этом заболевании. Как на Ваш взгляд, какие у нас шансы догнать своих сверстников в ПМР. Может надо параллельно еще какую-то терапию проводить? Ведь основные рефлексы у нас сохранились: ходит с поддержкой, садится за руки и сразу пытается встать (но сама, т.е. без чужих рук не встает), ест с ложечки, если даешь игрушку в руку, то берет (но взглядом за ними не следит), переворачивается.
Пожалуйста, дайте хоть какую-то информацию и статистику. А если есть возможность, то и заочную консультацию.
Большинство врожденных пороков центральной нервной системы представляют собой мультифакториальную патологию эмбрионального периода развития. Неврологическая симптоматика аномалий развития головного мозга зависит от их локализации и объема поражения. Кроме того, на степень неврологических расстройств может повлиять специфика патологической архитектоники тканей головного мозга и их соотношения между собой. Клиническая симптоматика этих аномалий малоспецифична. К наиболее частым неврологическим симптомам относят центральные парезы, эпилептические приступы, а также задержку психического и моторного развития различной степени выраженности. Среди наиболее часто встречаемых аномалий развития головного мозга выделяют кортикальные дисплазии, которые включают: фокальную кортикальную дисплазию, региональную и диффузную пахигирию, унилатеральную гемимегалэнцефалию, голопрозэнцефалию, шизэнцефалию, нейронные гетеротопии.
Фокальная корковая дисплазия – это очаговое нарушение нейронной миграции и дифференцировки. Выделяют несколько типов фокальных корковых дисплазий: тип 1, при котором нарушается корковая нейрональная организация при сохранности пирамидального рисунка коры, и тип 2, при котором имеет место выраженная дезорганизация с потерей пирамидального рисунка, при этом наблюдаются гигантские (баллонные) клетки . Основной локализацией фокальных корковых дисплазий является височная доля, наиболее эпилептогенная структура головного мозга. Агирия (лиссэнцефалия) – нарушение нейрональной дифференциации с уменьшением числа извилин вплоть до гладкого мозга. Характерен симптомокомплекс: микроцефалия, диффузная мышечная гипотония, эпилептические спазмы. Регионарная корковая дисплазия чаще представлена врожденным перисильвиарным синдромом. Суть нейроморфологических изменений заключается в билатеральной оперкулярной дисгирии. В клинической картине доминируют эпилептические приступы, псевдобульбарный и пирамидный синдром. Унилатеральная гемимегалэнцефалия – увеличение размеров одной доли или ее части вследствие избыточной пролиферации нейронов. Проявляется эпилептическими приступами, контрлатеральным гемипарезом . Голопрозэнцефалия – порок развития, при котором мозг остается нерасщепленным, часто сочетается с аномалиями лицевого скелета и приводит к летальному исходу в ранний постнатальный период. Шизэнцефалия проявляется «расщелинами» мозга, преимущественно в височной доле. В неврологическом статусе чаще наблюдаются резистентный эпилептический синдром, двигательные расстройства . Нейронные гетеротопии – нарушения нейрональной миграции на 35-й неделе гестации с образованием эктопированных участков нодулярной или ламинарной формы.
Согласно данным литературы, нейронные гетеротопии ответственны за 5-25% случаев эпилепсии у детей .
Наиболее показательным вариантом аномалии развития головного мозга является вариант ламинарной гетеротопии, когда слои гетеротипированных нейронов располагаются в глубоких и субкортикальных отделах головного мозга, известный как синдром «двойной коры».
Синдром «двойной коры» – это редкая, генетически обусловленная аномалия развития центральной нервной системы. Ее возникновение связано с мутацией гена даблкортина, локализованного в хромосоме Xg22, которая приводит к формированию ламинарной (ленточной) подкорковой гетеротопии нейронов. Вследствие подобного нарушения миграционных процессов создается иллюзия дублирования коры – «двойная кора» . Синдром впервые описан H. Jakob в 1936 г. и в дальнейшем выявлен S. Ricci и A Palmini у больных с эпилептическими синдромами . В клинической картине синдрома наиболее часто наблюдаются задержка психомоторного развития, терапевтически резистентная эпилепсия с преобладанием парциальных/астатических приступов и дебютом припадков преимущественно после 5 лет, четкие очаговые изменения на электроэнцефалограмме (ЭЭГ), могут также встречаться инфантильные спазмы в анамнезе. Лечение этого синдрома симптоматическое, основой которого является противоэпилептическая терапия .
Ниже приведен случай, отвечающий основным диагностическим критериям синдрома «двойной коры».
Клинический случай
Анамнез жизни и заболевания
Пациентка Г., 1995 г. р., родилась от четвертой беременности (1 – самопроизвольный аборт в ранние сроки, 2 – роды, здоровая дочь, 20 лет, 3 – медицинский аборт). Беременность протекала с угрозой прерывания на ранних сроках. Роды были срочные, физиологические. Масса при рождении составила 3200 кг, оценка по шкале Апгар – 8/8 баллов. Раннее моторное и речевое развитие проходило с некоторой темповой задержкой. В 5-летнем возрасте у нее появились серийные приступы «остановки» взгляда с замиранием, затем добавились фокальный компонент с тонической девиацией глаз влево и тонико-клонические подергивания в левой руке, далее – вторично генерализованные пароксизмы. Была проведена терапия фенобарбиталом и вальпроевой кислотой. В возрасте 10 лет у пациентки появились атонические, затем – аутомоторные приступы, к терапии добавили ламотриджин. Отмечено нарастание двигательных нарушений с формированием тетрапареза и когнитивных нарушений.
На момент поступления в неврологическое отделение (16.10.2012 г.) у больной сохранялись приступы потери сознания с падением без судорог, приступы миоклонических подергиваний головой с запрокидыванием длительностью от 3 до 5 минут, а также приступы «обмякания» со складыванием тела вперед. Частота пароксизмов в совокупности составила до 8-10 в сутки. Кроме того, отмечались жалобы на избыточную массу тела, косоглазие, снижение интеллекта.
Состояние при поступлении
При поступлении в стационар состояние пациентки по основному заболеванию классифицировалось как тяжелое. В неврологическом статусе: правая глазная щель была больше левой, зрачки равны, отмечено вертикальное косоглазие слева, сглажена правая носогубная складка, наблюдалась девиация языка и язычка влево. Мышечный тонус в конечностях дистоничен, без разницы сторон, движения в конечностях ограниченные, мышечная сила снижена в проксимальных отделах конечностей, сухожильные рефлексы равномерно оживлены, равны, патологические стопные знаки отмечаются с двух сторон, в пробе Ромберга отклонение назад и в стороны. Пальценосовую пробу выполняет с мимопопаданием. У больной имеет место лишний вес. Словарный запас и интеллект снижены.
Результаты обследования
Согласно данным нейропсихологического исследования, коэффициент интеллекта (IQ) пациентки соответствовал 62 баллам.
У больной был проведен ЭЭГ-мониторинг в течение 24 часов (аппарат электроэнцефалограф-регистратор «Энцефалан-ЭЭГр-19/86», производство «Медиком-мтд» г. Таганрог, Россия): во время бодрствования и ночного сна в лобных отведениях регистрировалась эпилептиформная активность в виде комплексов острая волна – медленная волна с тенденцией к генерализации (рис. 1).
Кроме того, была проведена магнитно-резонансная томография головного мозга (аппарат Hitachi Airis Mate 0,2 Тесла), согласно которой на аксиальных срезах определялись билатеральные лентовидные зоны, соответствующие серому веществу головного мозга, расположенные преимущественно субкортикально. Изгибы гетеротопированных слоев повторяли основную складчатость кортикальной поверхности. На коронарных срезах подтверждалось субкортикальное расположение гетеротопированных зон. В коре видимых диспластических изменений не отмечено. Таким образом, можно утверждать о наличии у пациентки МР-признаков билатеральной ламинарной гетеротопии серого вещества, что характерно для синдрома «двойной коры» (рис. 2).
 |
Обоснование диагноза и лечения
Таким образом, у больной отмечался ранний дебют эпипароксизмов со специфической динамикой и наслоением пароксизмов: фокальные – вторичная генерализация – астатические – аутомоторные пароксизмы, нарастающий когнитивный и неврологический дефицит, преобладание фокальной эпилептической активности на ЭЭГ и, наконец, наиболее значимый диагностический критерий – МР-признаки ламинарной гетеротопии серого вещества. В ходе обследования был выставлен диагноз: «аномалия развития центральной нервной системы: билатеральная ламинарная гетеротопия серого вещества головного мозга – синдром «двойной коры», эпилептическая энцефалопатия Леннокса – Гасто».
Пациентке была назначена противоэпилептическая терапия двумя препаратами – леветирацитамом в дозе 2000 мг/сут и ламотриджином по 200 мг/сут.
Катамнез в течение 6 месяцев показал купирование атонических приступов, но сохранение фокальных и аутомоторных. В перспективе возможна модификация противопилепической терапии: зонисамид, этосуксимид, лакосамид. Также обсуждается вопрос нейрохирургической коррекции для уменьшения количества пароксизмов.
Выводы
Рассмотренный случай подчеркивает необходимость придерживаться ряда облигатных принципов, ставших в ведущих эпилептологических центрах рутинными в повседневной практике врача-эпилептолога. К ним относятся такие принципы, как корректная синдромологическая диагностика пароксизмов, пролонгированный ЭЭГ-видеомониторинг, магнитно-резонансная томография высокой разрешимости по протоколу эпилептологического сканирования, генетическое типирование, что позволяет своевременно и точно диагностировать искомую патологию.
Использование магнитно-резонансной томографии является принципиально важным диагностическим инструментом для уточнения этиопатогенеза эпилепсии даже при наличии идиопатической ее формы. Трудно оценить всю значимость своевременного этиологического диагноза для выбора рациональной терапии, определения прогноза и семейного консультирования.
Литература
- Алиханов А.А. Нейрорадиологическая модель различных вариантов нарушения нейронной миграции // Журнал неврологии и психиатрии. – 2004. – № 10. – С. 81-85.
- Шестова Е.П., Евтушенко С.К., Соловьева Е.М., Душацкая А.В. Аномалии головного мозга (миграционные нарушения) у детей: клинико-радиологические проявления //Международный неврологический журнал. – 2005. – № 4 (4). – С. 30-36.
- Коновалов А.Н., Корниенко В.Н., Озерова В.И., Пронин И.Н. Нейрорентгенология детского возраста. – М.: Андор, 2001. – 456 с.
- Cohen M.M., Jr. The Child with Multiple Birth Defects / Second edition. – New York: Oxford University Press, 1997. – 267 p.
- Neil G. Epilepsy and Disorders of Neuronal Migration. I Introduction // Developmental Medicine and Child Neurology. – 1996. – V. 38. – Р. 1053-1057.
- Palmini A., Rim E-H., Da Costa J.C. Evidence for Focal accentuation if cortical dysfunction/excitability in the «Double cortex» syndrome // Epilepsy. – V. 38 (Suppl 3). – P. 6.
1 Детское клиническое территориальное медицинское объединение, г. Макеевка.
2 2 ООО « Медицинская лучевая диагностика», г. Макеевка.
Аномалии или мальформации головного мозга (лат. malus – негодный, плохой, дурной; греч. phorme – форма) – необратимые структурные дефекты, возникающие в результате нарушения нормального пре- или постнатального развития. Типы аномалий определяются временем патологического воздействия и его продолжительностью. Вероятными причинами аномалий являются генетические дефекты, внутриутробные инфекции (токсоплазмоз, сифилис, краснуха, цитомегаловирус, вирус простого герпеса, ВИЧ), лекарственное воздействие на плод, болезни матери в период беременности (диабет и другие метаболические нарушения), рентгеновское облучение на ранних этапах беременности, рецидивирующее маточное кровотечение, иными психоактивными веществами и др.
Нарушения развития нервной системы часто сопровождаются аномалиями костной системы и кожи. Некоторые аномалии возникают на 3-й неделе беременности. Так, неполное закрытие краниального конца нервной трубки приводит к анэнцефалии (летальный дефект). Дефектное закрытие каудальной части нервной трубки является причиной менингомиелоцеле. Пренатальная диагностика подобных нарушений возможна с помощью ультразвукового исследования. Известны следующие мальформации головного мозга.
1) Лиссэнцефалия (агирия ) и пахигирия (греч. lissos – гладкий; а – приставка отрицания; gyros – круг; pachys – толстый). При агирии кора головного мозга имеет очень мало извилин или они вообще отсутствуют, отсутствуют в ней и обычные клеточные слои. При пахигирии извилин мало (отсутствуют вторичные и третичные извилины), они необычно широки, имеют недостаточно организованное строение; борозды при этом выпрямлены, они короткие и неглубокие. В белом веществе встречаются гетеротопии нервных клеток. Клинически отмечаются тяжелая умственная отсталость, припадки, мышечная гипотония, сменяющаяся спастическим тетрапарезом. Чаще дети умирают в течение первого года жизни.
2) Полимикрогирия. При данной аномалии церебральная кора поделена на большое число очень маленьких складок, и ее поверхность принимает вид «сморщенного каштана». Обычно отмечаются тяжелая умственная отсталость и частые припадки, которые нередко резистентны к антиконвульсантам. Двусторонняя полимикрогирия оперкулярной области (расположенной под передней и задней центральными извилинами) проявляется псевдобульбарным синдромом Фуа -Шавани -Мари (выключением пирамидного влияния на 9–12-ю пары черепных нервов, ядра которых расположены в стволе мозга). При этом, в отличие от других вариантов псевдобульбарного синдрома, очень редко возникают приступы насильственного смеха.
3) Кортикальная дисплазия . Характеризуется локальной аномалией коры в виде гигантских нейронов и астроцитов, а также хаотическим расположением корковых слоев. Патология выявляется на МРТ. Обычно наблюдаются нарушения интеллектуального развития, парциальные эпилептические припадки, миоклонии, легкий гемипарез.
4) Агенезия мозолистого тела . Может быть частичной или полной (в последнем случае 3-й желудочек мозга остается открытым). Обычно отсутствует задняя часть мозолистого тела, так как его рост идет спереди назад (эта часть соединяет в основном затылочные доли мозга). Частота данной аномалии в точности не установлена (от 5 на 7000 и более). Агенезия мозолистого тела может быть изолированным дефектом либо сочетается с другими аномалиями ЦНС и иных органов. Причиной каллезной агенезии могут быть генетические дефекты и/или метаболические нарушения.
Когда агенезия мозолистого тела сочетается с изменениями сетчатки, аномалиями глазных яблок, позвоночника и сосудистых сплетений желудочков мозга, микроцефалией, умственной отсталостью (до степени идиотии), нистагмом и младенческими судорогами (спазмами сгибательной мускулатуры, миоклоническими приступами), этот симптомокомплекс называется синдромом Айкарди . Развивается он только у девочек вследствие дефекта одной из хромосом Х (мальчики с такой хромосомой погибают до рождения).
В свою очередь синдром Шапиро , помимо признаков агенезии, проявляется гипоталамической дисфункцией, в частности гипотермией. Изолированная агенезия может протекать бессимптомно и обнаруживается случайно при КТ и МРТ. Однако у 2/3 пациентов с агенезией выявляется эпилепсия, а у половины из них обнаруживается нарушение интеллектуального развития.
5) Порэнцефалия (греч. porus – дыра). Характеризуется наличием одной или нескольких полостей в большом мозге, возникающих либо внутриутробно, либо в раннем постнатальном периоде. Происходит это вследствие травмы мозга, нейроинфекции, кровоизлияния и особенно часто ишемии мозга. Порэнцефалические кисты ишемической этиологии обычно расположены в бассейне средней мозговой артерии (при одностороннем поражении в 80% случаев они располагаются слева). Подобная ишемия может быть следствием неправильного развития сосудов, возникать из-за их спазма (например, если мать во время беременности принимала кокаин), эмболической окклюзии сосуда плацентарными фрагментами или тромбоза при дегидратации и ДВС-синдроме. Порэнцефалия всегда предполагает сообщение с субарахноидальным пространством, а часто – и с желудочковой системой.
Большинство порэнцефалических кист не нуждается в лечении, но иногда они расширяются и ведут к повышению внутричерепного давления из-за образования клапанного механизма, препятствующего оттоку ликвора из кисты. В таких случаях проводится шунтирующая операция. Различают истинную и ложную порэнцефалию. В первом случае полости выстланы эпендимой и сообщаются с вентрикулярной системой и субарахноидальным пространством. При ложной порэнцефалии полости замкнуты, лишены эпендимной выстилки и возникают вследствие энцефаломаляции разного происхождения. Нарушения психического развития возникают во всех случаях, если киста имеет значительные размеры, со временем увеличивается или их бывает несколько.
6) Микроцефалия, или синдром Джакомини, встречается у 10–11% пациентов с умственной отсталостью. Частота микроцефалии – один случай на 5000 новорожденных. Соотношение массы мозга и массы тела при рождении в типичных случаях микроцефалии составляет 1:100 (в норме оно равно в среднем 1:8). Микроцефалия проявляется уменьшенной окружностью головы, дефицит составляет более чем 5 см от средних показателей. Типично и дальнейшее отставание роста мозгового черепа (микрокрания). При этом швы черепа могут долго оставаться открытыми. Кости черепа у пациентов часто утолщены, в них рано формируются диплоидные каналы. Внутричерепное давление обычно не повышено. Строение головного мозга характеризуют недоразвитие и неправильная структура больших полушарий при сравнительно нормальной цитоархитектонике мозжечка и ствола мозга. Пациенты отстают от сверстников не только в психическом, но и в физическом развитии. В зависимости от причин развития различают следующие варианты микроцефалии.
1. Истинная микроцефалия . Носит наследственный характер, передается по аутосомно-рецессивному и сцепленному с Х-хромосомой типам. Описаны семейные случаи расстройства, а также формы с аутосомно-хромосомной патологией. Среди микроцефалов эта форма встречается в 7–34% случаев. В родословной пациентов часто встречаются лица с умственной отсталостью, уменьшенными размерами черепа, невысоким интеллектом, эпилептическими припадками. В наследственно-рецессивных формах микроцефалии у 1/8 пациентов выявляются близкородственные браки. Распространенность гена микроцефалии составляет, по разным данным, 1 на 180–230 человек. Умственная отсталость чаще бывает тяжелой (идиотия) и глубокой (имбецильность) степени, реже встречается умеренная и легкая умственная отсталость.
2. Эмбриопатическая или вторичная микроцефалия . Возникает вследствие нарушений развития в пренатальном периоде по разным причинам, таким как нейроинфекции (грипп, токсоплазмоз, краснуха, цитомегаловирусный энцефалит), интоксикации (алкоголь, наркотики, промышленные отравления), внутриутробная травма или асфиксия, нарушение витаминного баланса, алиментарный дефицит, нарушения обмена фосфора, кальция. При вторичной микроцефалии в головном мозге нередко обнаруживают кисты, очаги кровоизлияния или обызвествления.
3. Синдромологические варианты микроцефалии . Развиваются в результате различных хромосомных аберраций (трисомии, моносомии, делеции хромосом, инверсии фрагментов хромосом, кольцевые хромосомы, транслокации хромосом). В 25% это патология аутосом; в 25% – половых хромосом, в основном Х-хромосомы; в 50% – различные изменения структуры различных аутосом.
7) Макроцефалия . Характерны увеличение массы и объема головного мозга, а вместе с этим и мозгового черепа при рождении. Встречается значительно реже микроцефалии. В большинстве случаев сопровождается нарушением расположения мозговых извилин, изменениями структуры коркового слоя полушарий, очагами гетеротопии в белом веществе (наличием нейронов, которые не успели мигрировать в сторону коры). Костные швы не расширены, желудочки мозга нормального или почти нормального размера. При макроцефалии часто наблюдается умственная отсталость разной степени выраженности, иногда – судорожные припадки. Причинами макроцефалии могут быть поражения ткани мозга вследствие нарушения липидного обмена.
Встречается частичная макроцефалия – увеличение одного из полушарий. Обычно она сочетается с асимметрией мозгового черепа. В части случаев парциальная макроцефалия является результатом неопухолевого объемного процесса в средней черепной ямке (гематома, гигрома, кистозный арахноидит и др.).
8) Менингоэнцефалоцеле . Церебральная грыжа встречается реже, чем спинальная. В половине случаев церебральной грыжи обнаруживается сопутствующая гидроцефалия. Не менее часты соматические уродства. При истончении стенок грыжи возможно истечение ликвора и развитие менингита. При маленьких грыжах, которые не содержат ткань мозга, показано хирургическое лечение и закрытие кожного дефекта. При грыжах, которые содержат ткань мозга, особенно при их большом объеме, прогноз даже при хирургическом лечении плохой. Исход более благоприятен при передних, чем при задних мозговых грыжах.
9) Сирингомиелия (греч. syrings – трубка, полость; myelos – спинной мозг). Врожденное хроническое заболевание, для которого характерны: а) расширение центрального канала спинного мозга, заполненного ликвором (гидромиелия); б) формирование вокруг центрального канала полостей и разрастаний нейроглии в сером веществе спинного мозга, чаще в шейном и верхнегрудном отделах. Если такие изменения появляются в нижней части ствола мозга, то говорят о сирингобульбии . Нередко сирингомиелия сочетается с врожденной гидроцефалией. Распространенность болезни, по разным данным, составляет от 0,3 до 7,3 на 100 000 населения. Существует связь сирингомиелии с этнической идентичностью. Сообщается, что у татар в Башкирии она встречается с частотой 130 на 100 000 и что марийцы болеют в 7 раз чаще башкир и в 13 раз чаще русских. Заболевание чаще наблюдается у мужчин в северных широтах, особенно занятых тяжелым физическим трудом.
Природа заболевания окончательно не установлена, существует ряд теорий его развития, среди которых основной считается дизонтогенетическая (греч. dys – приставка, указывает на расстройство; ontos – бытие, сущее; genesis – развитие, происхождение). В соответствии с возможными причинами развития различают несколько вариантов сирингомиелии: 1) связанная с аномалией развития задней черепной ямки (аномалия Киари 1-го типа – самая частая находка, базиллярная импрессия, арахноидальные кисты в области большой затылочной цистерны); 2) посттравматическая сирингомиелия; 3) сирингомиелия вследствие спинального менингита и арахноидита; 4) сирингомиелия, сопутствующая опухолям спинного мозга; 5) идиопатическая сирингомиелия, не связанная с вышеперечисленной патологией.
Ведущей патогенетической концепцией сирингомиелии является теория W.J. Gardner (1950), согласно которой к болезни приводят ликвородинамические удары из 4-го желудочка в стенки центрального канала спинного мозга с последующим его расширением.
Первые признаки сирингомиелии по данным появляются на 3–6-й неделе внутриутробного развития, в период формирования нервной трубки. Явные клинические проявления обнаруживаются на 2–3-м десятилетии жизни. Обычно это нарушения болевой и температурной чувствительности, чаще на шейно-грудном уровне. С этим связаны многочисленные безболевые травмы и ожоги пациентов. Тактильная и мышечно-суставная чувствительность страдает реже и в меньшей степени. Типичным признаком болезни является резкая приступообразная или постоянная жгучая боль, локализованная соответственно пораженным сегментам спинного мозга. Обычно выявляются и вегетативно-трофические расстройства: отеки, де- или гиперпигментация кожи, кожные рубцы, омозолелость кожи, трофические язвы, мышечные атрофии, гангрена и мутиляция (лат. mutilis – увечный, искалеченный) концевых фаланг, панариции, деформации костей и мн. др. Нередко выявляется дизестезия – искажение чувствительности, когда, например, болевое раздражение воспринимается как ощущение холода или жжения.
При вовлечении в процесс ствола мозга возникают нарушения со стороны черепных нервов (параличи мимических и жевательных мышц, наружной прямой мышцы глаза, парез мягкого неба, глотки и голосовых связок, асимметричная атрофия мышц языка, нистагм, головокружение и др.). Из психиатрической патологии у части пациентов отмечаются задержка психического развития, нарушения социальной адаптации, депрессия. У пациентов и членов их семей могут наблюдаться разнообразные сочетанные аномалии: деформации грудной клетки, кифосколиоз, непропорционально длинные руки, искривление пальцев, аномалии роста волос, строения ушей и т. д.
10) Мальформации Киари . Представляют собой дисгенезию мозжечка в сочетании с широким кругом аномалий ромбовидного, среднего и межуточного мозга. Различают ряд вариантов данной аномалии. Чаще других встречается «взрослый тип» аномалии в виде одно- или двустороннего опущения мозжечковых миндалин через большое затылочное отверстие в позвоночный канал. Клинические проявления (стволовые и мозжечковые симптомы) возникают только на 3–4-м десятилетии жизни.
«Детский тип » аномалии Киари представляет собой смещение мозжечка, ствола и 4-го желудочка вниз через большое затылочное отверстие. При этом всегда наблюдается конгенитальная гидроцефалия, часто – стеноз водопровода мозга. Уже при рождении выявляется связанная с врожденной водянкой головного мозга симптоматика (увеличение окружности головы, расхождение черепных швов, выбухание родничков и др.). При мальформации Киари 3-го типа мозговая грыжа включает мозжечок и в половине случаев – затылочную долю. Лечение хирургическое.
11) Синдром Денди -Уокера . Встречается с частотой 1 на 25 000–30 000. Ген данной аномалии локализован на хромосоме 3, большинство случаев болезни является спорадическим. Характерно частичное или полное отсутствие мозжечкового червя, кистозное расширение 4-го желудочка, увеличение задней черепной ямки и раннее развитие гидроцефалии. Сопутствующие аномалии большого мозга и задержка умственного развития отмечаются в 70%, пороки внутренних органов – в 20–80% случаев. УЗИ-диагностика возможна с 18–20 недель беременности.
12) Арахноидальные кисты . Представляют собой заполненные жидкостью полости, возникающие при удвоении паутинной оболочки или расположенные между arachnoidea и мягкой мозговой оболочкой. Они могут сообщаться или не сообщаться с субарахноидальным пространством. Частота кист по данным КТ и МРТ – 4%. Кисты располагаются в сильвиевой щели (50%), мостомозжечковом углу (10%), четверохолмии (10%), супраселлярной области (10%), черве мозжечка (8%), на конвекситальной поверхности мозга (5%) или имеют иную локализацию (7%). Клинические симптомы присутствуют только в 20–30% случаев (даже огромные кисты могут быть бессимптомными).
При срединном расположении кисты могут сдавливать водопровод мозга или отверстие Монро, вызывая обструктивную гидроцефалию. Кроме признаков внутричерепной гипертензии и гидроцефалии возможны задержка психического развития, зрительные нарушения, параличи черепных нервов, атаксия, припадки, эндокринные расстройства и деформация черепа. Лечение хирургическое (при наличии серьезной и прогрессирующей клинической симптоматики), симптоматическое. При обструктивной гидроцефалии проводят шунтирование как кисты, так и желудочковой системы.
13) Синдром Мебиуса . Врожденное двустороннее поражение лицевого и отводящего нервов (аплазия ядер, самих нервов либо иннервируемых ими мышц). Встречаются как наследственные, так и спорадические случаи. Описано сочетание синдрома Мебиуса с атрофией большой грудной мышцы, косолапостью и умственной отсталостью.
14) Анэнцефалия . Отсутствие большого мозга, костей свода черепа и покрывающих его мягких тканей. На месте мозгового вещества обычно располагаются соединительная ткань, богатая кровеносными сосудами, с кистозными полостями, выстланными медуллярным эпителием (клетками, выстилающими полости мозга; лат. medullia – костный мозг), глиальная ткань, единичные нервные клетки, остатки сосудистых сплетений.
15) Эксэнцефалия . Отсутствие костей черепа (акрания), а также мягких покровов головы. Большие полушария располагаются открыто на основании черепа в виде отдельных узлов, покрытых мягкой мозговой оболочкой.
16) Гидроанэнцефалия . Полное или почти полное отсутствие больших полушарий, в то же время кости черепа и его покровные ткани сохранены. Голова при этом нормальной величины или несколько увеличена. Полость черепа заполнена главным образом цереброспинальной жидкостью. Продолговатый мозг и мозжечок достаточно развиты, средний мозг и другие отделы головного мозга могут отсутствовать или представлены рудиментарно.
17) Прозенцефалия (греч. pros – по направлению к). Редкая аномалия (1:16 000), при которой большие полушария разделяет лишь мелкая продольная борозда.
18) Голопрозенцефалия (греч. holos – весь). Аномалия, при которой большие полушария и боковые желудочки не разделены. Часто бывают и иные пороки (нарушения строения лица и его костей, циклопия и др.). Обычно смерть наступает вскоре после рождения, дети с циклопией рождаются мертвыми. В части случаев данной аномалии обнаруживается трисомия хромосом 13–15.
19) Болезнь Луи -Бар (1941), или атаксия-телеангиэктазия. Наследуется по аутосомно-рецессивному типу с высокой пенетрантностью мутантного гена. У детей, начинающих ходить, обнаруживается нарастающая мозжечковая атаксия, позднее появляются гиперкинезы (атетоз, миоклонии), сухожильная гипорефлексия, дизартрия, могут быть поражения черепных нервов, затруднения произвольных движений глаз (окуломоторная апраксия). На коже лица и шеи, конъюнктиве у детей в возрасте 3–6 лет появляются симметричные телеангиэктазии, обычно распространяющиеся затем на мозговые оболочки, вещество мозга. В связи с генетически обусловленным клеточным и гуморальным иммунитетом выявляется склонность к хроническим воспалительным заболеваниям (пневмонии, синуситы, бронхиты, тонзиллиты и др.).
К 12–15 годам возникают нарушения глубокой и вибрационной чувствительности. Позднее в связи с поражением клеток передних рогов спинного мозга развиваются атрофии мышц, обнаруживаются фасцикулярные подергивания. На коже появляются пигментные пятна кофейного цвета, участки гипопигментации, себорейный дерматит, поседение волос. Характерна задержка психического и физического развития. Обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммаглобулинемия, поражение ретикулоэндотелиальной системы (ретикулемы, лимфосаркома и др.). Прогноз плохой. Причиной смерти являются хронические заболевания бронхов и легких, лимфомы, карциномы.
20) Гипомеланоз Ито . Обычно описывается как спорадическое заболевание, хотя имеются единичные случаи с аутосомно-доминантным и аутосомно-рецессивным типами наследования. По частоте уступает лишь нейрофиброматозу и туберозному склерозу. Поражает оба пола, однако у женщин встречается в 2,5 раза чаще, чем у мужчин.
При гипомеланозе нарушена миграция клеток из нервной трубки зародыша: меланобласты мигрируют к месту своего назначения во второй половине беременности, а нейроны мигрируют из ганглионарной пластинки между 3-м и 6-м месяцами эмбрионального развития. Этим объясняют гетеротопию серого вещества головного мозга и малое число меланоцитов в мальпигиевом слое кожи.
Кожные проявления болезни сводятся к участкам гипопигментации причудливой формы, которые обнаруживаются при рождении или появляются в первые месяцы после родов. После полового созревания они могут полностью исчезать. Могут иметь место также другие кожные дисплазии: сосудистые невусы, «монголоидные» голубые пятна, пятна цвета кофе с молоком, невус Ота и др.
Гетеротопия серого вещества головного мозга проявляется умственной отсталостью (92% случаев), часто встречаются аутизм, двигательная расторможенность. Более чем в половине случаев отмечаются судорожные припадки, резистентные к антиконвульсантам, нередко – диффузная мышечная гипотония. У четверти пациентов наблюдается макроцефалия, реже – микроцефалия, нередки другие аномалии (внутренних органов, гениталий и др.). Лечение симптоматическое.
21) Синдром Блоха -Сульцбергера, или недержание пигмента. Предполагается доминантное Х-сцепленное наследование, которое часто оказывается летальным уже в пренатальном периоде для пораженных гемизигот (мужчин). Предполагается также, что заболевание может быть обусловлено одним из двух генов: первый из них картирован на коротком плече Х-хромосомы (Хр11.21), второй – на длинном плече этой хромосомы (Хq28). Женщины болеют в 20 раз чаще мужчин.
В первые полгода у детей появляется линейно расположенная буллезная и везикулезная сыпь на коже туловища и конечностей, в пузырном содержимом обнаруживается много эозинофилов. В начале болезни эозинофилия отмечается и в крови (до 50%). Позднее возникают лихеноидные и гиперкератозные высыпания, также располагающиеся преимущественно линейно. После регресса воспаления или несколько ранее возникают пигментные изменения кожи сероватого или голубоватого оттенка в виде паутин, полос, завитков – это массивные отложения пигмента, который из базального слоя кожи перемещается в меланофаги верхней части кожи (отсюда термин «недержание пигмента»). К подростковому возрасту они могут исчезать. Наблюдаются и другие изменения кожи и ее придатков: ангидроз, алопеция, дистрофия волос, зубов, ногтей.
Наблюдается, кроме того, церебральная симптоматика, в первую очередь умственная отсталость (1/3 случаев), эпилептические припадки (до 5%случаев). Обычно бывают гидроцефалия, микроцефалия, медленно прогрессирующая слабость в конечностях со спастикой или мышечной гипотонией. У трети пациентов выявляются атрофия зрительных нервов, папиллит, ретинальная пигментация, страбизм, нистагм и катаракта. На МРТ у пациентов с психиатрической и неврологической симптоматикой обнаруживаются гипоплазия мозолистого тела, глиоз, энцефаломаляция и фокальная атрофия полушария головного мозга и мозжечка в гемисфере, контралатеральной стороне с более выраженными кожными изменениями. Лечение симптоматическое.
22) Синдром эпидермального невуса . Чаще встречаются спорадические случаи, описаны, однако, семьи с аутосомно-доминантным типом наследования. Характеризуется комбинацией эпидермального невуса (большей частью расположенного на лице, реже – на скальпе) и аномалий развития мозга, а также глаз, скелета, почек или сердца. Гистологически невус характеризуется гиперкератозом, папилломатозом и акантоцитозом. На коже могут быть также участки гиперпигментации, пятна цвета кофе с молоком и другие невусы.
Изменения в мозге: гемимакроэнцефалия, гиперплазия и гипертрофия нейронов и глии в гемисфере, ипсилатеральной невусу. Клинически это проявляется умственной отсталостью, чувствительными к антиконвульсантам припадками и контралатеральным гемипарезом. Вторичные поражения ЦНС, вызванные сосудистой патологией (ангиомы, коарктация аорты, отсутствие синусов твердой мозговой оболочки), включают инфаркты, атрофию, порэнцефалию, гидроцефалию и кальцификаты. Лечение симптоматическое.
23) Существуют и иные редкие нейрокожные синдромы с наследственной и неизвестной этиологией, при которых наблюдаются умственная отсталость, задержка психического развития, речевые и другие нарушения, припадки: синдромы Горлина (базальноклеточный невус), де Сантиса-Какьоне (ксеродермическая идиотия), Руда, Ван-Богарта-Диври (диффузный кортикоменингеальный ангиоматоз) и некоторые другие.
24) Детский церебральный паралич (ДЦП). Характеризуется не прогрессирующими церебральными двигательными и иными нарушениями, возникающими на ранних этапах жизни вследствие поражения ЦНС под влиянием различных причин. При этом английский хирург Литтл, который впервые описал в 1860-х годах спастическую диплегию (именуемую болезнью Литтла), положил начало широко распространенному ошибочному предположению, будто причиной развития ДЦП является гипоксия в родах. З. Фрейд (1897), в то время еще невролог и автор монографии «Инфантильный церебральный паралич», первым обратил внимание на такие нарушения при ДЦП, как умственная отсталость, эпилептические припадки и нарушения зрения.
Частота ДЦП, по разным данным, составляет 2,5–5,9 на 1000 новорожденных. По сведениям К.А. Скворцовой (1994), ДЦП является причиной 24% случаев детской неврологической инвалидности.
Причинами развития ДЦП могут быть генетические дефекты (наследственные дисгенезии головного мозга являются причиной 10–12% всех случаев спастических форм ДЦП), известные перинатальные инфекции (краснуха, цитомегалия, грипп, токсоплазмоз и др.) и токсикозы матери во время беременности, нейровизуализируемые аномалии развития головного мозга, атрофические процессы и избыточное количество врожденных микроаномалий, выраженная и длительная асфиксия в дородовом периоде и в родах, кровоизлияние в мозг, травмы и болезни (энцефалит, менингоэнцефалит, гнойный менингит) у ребенка в раннем постнатальном периоде, гипоксически-ишемическая энцефалопатия. Число случаев ДЦП, связанного в своем развитии с внутриродовой асфиксией и травмой, составляет около 10%.
Одним из самых надежных предикторов ДЦП является перивентрикулярная лейкомаляция у недоношенных детей – ДЦП выявляется в 22–100% случаев ее обнаружения. Перивентрикулярная лейкомаляция расценивается как гипоксически-ишемическое повреждение за счет свойственной детям с этой параклинической находкой нарушений мозгового кровообращения, в частности артериальной гипотонии у младенцев. Согласно другой гипотезе, очаги лейкомаляции возникают за счет воздействия цитокинов, освобождающихся при внутриутробных инфекциях, ускоряющих роды в начале 3-го триместра беременности.
В целом, по мнению большинства исследователей, в развитии ДЦП преобладает роль наследственных и конституциональных факторов, на которые интранатальные и перинатальные воздействия чаще всего лишь наслаиваются и служат отягощающим обстоятельством. Однако лишь в относительно немногих случаях удается установить специфическую этиологию (генетические синдромы, врожденные мальформации и внутриутробные инфекции ЦНС). У большинства пациентов с ДЦП удается идентифицировать лишь факторы риска, актуальные во время беременности, перед нею или во время перинатального периода.
К пре- и интранатальным факторам риска развития ДЦП (указывающим на более высокую вероятность появления расстройства) относят следующие:
1) два и более предшествующих аборта;
2) большое количество предыдущих родов;
3) кровотечение во время беременности;
4) признаки преэклампсии;
5) меньшая окружность головы при рождении;
6) короткая пуповина и/или приращение плаценты;
7) низкая для срока рождения масса тела ребенка (менее 2001,0 г). Так, у младенцев, рожденных с массой тела менее 1300,0 г, частота ДЦП в 20–25 раз выше, чем у имеющих нормальную массу тела детей. Дети, рожденные с массой менее 2500,0 г, составляют около трети всех детей, у которых в последующем развивается ДЦП.
Факторами риска считаются также большие интервалы между менструациями у матери ребенка, аномально короткие (до 3 месяцев) либо необычно длительные (более 3 лет) интервалы с момента предыдущих родов беременности, наличие в анамнезе матери спонтанных абортов или мертворожденных, гипербилирубинемия плода, артериальная гипотония у младенцев. Близнецы более часто страдают ДЦП. Родоразрешение путем кесарева сечения в таких случаях не приводит к снижению частоты ДЦП. Повышает риск рождения ребенка с ДЦП прием эстрогенов и тиреоидных гормонов матерью, наличие у матери гипертиреоза, хорионита.
Постнатальные факторы, такие как менингоэнцефалит, черепно-мозговая травма, окклюзия церебральных сосудов, рассматриваются как причины 12–21% всех случаев ДЦП. Тщательное исследование таких пациентов выявляет в ряде случаев признаки более раннего патологического пренатального влияния на плод (микродисгенезии головного мозга, большое количество других микроаномалий, короткая пуповина и др.).
Патогенез ДЦП представляют следующим образом. Патогенные факторы, действующие во время эмбриогенеза, вызывают разнообразные и достаточно серьезные аномалии развития мозга. На более поздних этапах внутриутробного развития (в период фетогенеза) возможны замедление процессов миелинизации нервной системы, нарушение дифференциации нервных клеток, патология формирования межнейрональных связей и сосудистой системы мозга. При несовместимости крови матери и плода по резус-фактору, системе АВО и другим антигенам эритроцитов в организме матери вырабатываются антитела, вызывающие гемолиз эритроцитов плода. Непрямой, не связанный с белком билирубин, образовавшийся в процессе гемолиза, оказывает токсическое воздействие на нервную систему, в частности на структуры стриопаллидарной системы.
У плодов, перенесших внутриутробную гипоксию, к моменту рождения защитные и адаптационные механизмы оказываются недостаточно сформированными, что способствует развитию родовой черепно-мозговой травмы и асфиксии во время родов. В патогенезе поражений нервной системы, развивающихся во время родов и постнатально, главную роль играют гипоксия плода, ацидоз, гипогликемия и другие метаболические нарушения, ведущие к отеку мозга и вторичным расстройствам мозговой гемодинамики и ликвородинамики.
Существенное значение в патогенезе ДЦП придается иммунопатологическим процессам: мозговые антигены, образующиеся при деструкции нервной ткани под влиянием инфекций, интоксикации, механических повреждений мозга плода, могут привести к появлению соответствующих антител в крови матери, что негативно сказывается на развитии мозга плода.
Систематики ДЦП, основанной на знании его этиологии, патогенеза или времени повреждения церебральных структур, не существует. Принятые ныне базируются на характере клинической симптоматики расстройства.
1. Наиболее часто встречаются спастические формы ДЦП – 79% всех случаев данной патологии. Они могут быть представлены как симметрическими, так и асимметрическими нарушениями центральных мотонейронов (пирамидных нейронов передней центральной извилины – коркового центра произвольной моторики). Спастические формы ДЦП представлены в трех вариантах:
а) спастическая диплегия (синдром Литтла) – самый частый вариант ДЦП, особенно у недоношенных детей (составляет 41% всех случаев ДЦП). Характеризуется тетрапарезом с вовлечением в патологический процесс также мышц лица, языка, глотки. При этом наиболее выражены двигательные расстройства в нижних конечностях – нижний спастический парапарез со спастикой приводящих мышц бедер и мышц-разгибателей. Если ребенок лежит, ноги у него вытянуты, а при попытке его поставить ноги перекрещиваются, и он опирается не на всю стопу, а только на переднюю ее часть.
В связи с постоянным напряжением приводящих мышц бедер ноги слегка согнуты в тазобедренных и коленных суставах и ротированы внутрь. При попытке ходить с посторонней помощью ребенок совершает танцующие движения, поворачивая тело в сторону ведущей ноги. Нередко выраженность парезов асимметрична, при этом различия в возможностях активных движений особенно отчетливо проявляются в руках.
У значительной части детей отмечается лишь легкая неловкость в руках и при адекватном обучении ребенок может научиться писать, рисовать, обслуживать себя, овладеть мануальными практическими и трудовыми навыками.
На фоне диплегии возможны хореатетоидные гиперкинезы, в которые вовлекаются прежде всего мимические мышцы и мышцы дистальных отделов рук. Если расстройство сочетается с поражением околожелудочкового белого вещества во время родов, то оно может осложниться гидроцефалией. У большинства пациентов обнаруживаются расстройства развития речи (артикуляции, экспрессивной, импрессивной речи и др.), задержка психического развития, инфантилизм, а также признаки органического снижения психической деятельности (повышенная утомляемость, замедление темпа психических процессов, снижение памяти, недостаточная концентрация внимания, эмоциональная неустойчивость), явления вегетодистонии (потливость, бледность кожных покровов, нарушения терморегуляции и т. п.).
Дети с развитым интеллектом начиная с 2–3-летнего возраста обычно тяжело переживают наличие двигательных нарушений, поскольку они препятствуют активному исследованию внешнего мира и взаимодействию с ним. Они подавлены, не уверены в себе, редко бывают жизнерадостными, неохотно вступают в контакт со здоровыми детьми, общение с которыми обостряет чувство своей неполноценности. Лучше они чувствуют себя в обществе детей с подобными нарушениями, где не так болезненно ощущают свою ущербность;
б) двойная диплегия, или квадриплегия, встречается у 19% детей с ДЦП. Характеризуется тетрапарезом, при котором руки страдают в большей степени, чем ноги, или они поражены приблизительно в равной степени. Возможна асимметрия выраженности парезов. При этом тонус мышц высокий, имеется сочетание спастики и ригидности (поражения пирамидной и экстрапирамидной систем) обычно с преобладанием ригидности. Реакции равновесия развиты недостаточно. Почти всегда выражены элементы псевдобульбарного паралича, в связи с чем затруднены жевание, глотание, речь. Нередки судорожные пароксизмы, микроцефалия. При этой форме встречаются наиболее значительные проявления умственного недоразвития. На КТ и МРТ чаще всего обнаруживаются множественные кисты в белом веществе больших полушарий, а также полости, сообщающиеся с боковыми желудочками. Выявляются, кроме того, диффузная атрофия коры большого мозга и гидроцефалия;
в) спастическая гемиплегия составляет 19% случаев ДЦП. Характеризуется соответствующими двигательными нарушениями преимущественно на одной стороне. При этом нередко двигательные расстройства более выражены в руке – она согнута во всех суставах, кисть у детей раннего возраста сжата в кулак, в более позднем возрасте имеет форму «руки акушера». Нередко возникают фокальные эпилептические припадки по типу двигательного Джексона. Интеллектуальное развитие обычно является близким к нормальному.
Наиболее частой находкой у пациентов с гемиплегической формой ДЦП при патоморфологическом исследовании или нейровизуализации (КТ, МРТ) является атрофия в зоне кровоснабжения средней мозговой артерии. Генез этих нарушений (геморрагия или ишемия) установить до последнего времени не удалось. По неясным причинам правосторонняя гемиплегия встречается в 2 раза чаще левосторонней (это указывает на повреждение преимущественно доминантной гемисферы, что косвенно свидетельствует о том, что латерализация мозговых функций происходит в основном в период внутриутробного развития и является врожденной). У некоторых детей с гемиплегической формой ДЦП выявляется перивентрикулярная атрофия (атрофия белого вещества, располагающегося вокруг желудочков мозга), а в 1/6 случаев – значительные мальформации развития мозга.
У 1/3–1/4 пациентов с конгенитальной гемиплегией данные КТ и МРТ не выявляют какой-либо патологии. На этом основании предполагают, что ряд случаев ДЦП связан с ранними аномалиями развития головного мозга на микроскопическом уровне (тем самым значительно снижается вероятность гипотезы, согласно которой ДЦП обусловлен повреждением нормально развивающегося мозга). Подтверждает предположение о роли невизуализируемых и ранних изменений мозга также то, что, по данным некоторых исследований, в акушерском анамнезе большинства пациентов с гемиплегической формой ДЦП не выявляется какой-либо клинически значимой патологии.
2. Дискинетические формы ДЦП встречаются приблизительно в 10% случаев данной патологии. Предполагается, что они развиваются вследствие иммунной несовместимости крови плода и матери, встречаясь в основном у доношенных детей. Психическое развитие страдает при них меньше, чем при других формах ДЦП, но часто встречается дизартрия. Дискинетические ДЦП представляют два варианта расстройства:
а) атетоидная или гиперкинетическая форма ДЦП характеризуется повреждением преимущественно стриопаллидарной системы, а клинически – насильственными движениями, по большей части хореатетозом, атетозом, хореей, которые преобладают в мышцах шеи, мимических мышцах и мышцах проксимальных отделов конечностей. Гиперкинезы обычно провоцируются началом движения в одной конечности и распространяются затем с мышц этой конечности на другие мышечные группы. Микроскопическое исследование базальных ганглий часто обнаруживает в них мраморный рисунок (status marmoratus);
б) дискинетическая форма ДЦП характеризуется, во-первых, тем, что при активных движениях возникают аномальные изменения тонуса во многих мышечных группах с развитием патологических поз (обычно стереотипных). Дистония иногда ошибочно принимается за спастику. В сомнительных случаях прибегают к исследованию пациента в положении лежа на спине: в этой позиции у больного со спастикой тонус не изменяется, а у больного с дистонией выявляется пониженный тонус.
Во-вторых, это форма ДЦП со стойкой мышечной гипотонией, связанная с поражением преимущественно церебеллярных путей, связывающих мозжечок с выше- и нижерасположенными нервными структурами, контролирующими движения. Мышечная гипотония при этом часто сопровождается пирамидной симптоматикой (оживление глубоких рефлексов, патологические стопные рефлексы). На КТ головного мозга в таких случаях наиболее частой находкой оказывается внутренняя гидроцефалия (расширение желудочковой системы). Данный вариант в некоторых классификациях относится к мозжечковому ДЦП.
3. Мозжечковые формы ДЦП встречаются в 10–11% случаев данной патологии. Это простая атактическая форма ДЦП и атактическая диплегия. В первом случае наблюдается статическая и динамическая атаксия (нарушения стояния и ходьбы), обусловленная главным образом поражением мозжечка и его связей. Могут быть другие мозжечковые симптомы (нистагм, адиадохокинез, дисметрия и т. п.). Во втором случае атаксия сочетается с атонически-астатическим синдромом, признаками умеренного спастического пареза в связи с вовлечением в процесс корково-подкорковых структур мозга.
Существуют, наконец, многочисленные смешанные формы ДЦП, при которых выявляются различные комбинации повреждения мозговых структур, разграничения здесь достаточно условны.
Помимо двигательных при ДЦП наблюдаются другие нарушения. Судорожный синдром, умственное недоразвитие, затруднения в учебе, нарушения зрения и слуха, страбизм, дизартрия встречаются при ДЦП с существенно большей частотой, чем в детской популяции в среднем.
Эпилептический синдром наблюдается при ДЦП в 35% случаев. Наиболее часто он развивается при гемиплегической форме ДЦП. Обычно припадки возникают в первые 2 года жизни. Наличие припадков и умственной отсталости при этой форме ДЦП коррелирует с выраженностью выявляемых при нейровизуализации церебральных нарушений. У пациентов с дискинетической формой ДЦП припадки встречаются сравнительно редко, как и при атактической форме.
У больных с гемиплегической формой ДЦП часто обнаруживается гомонимная гемианопсия (выпадение поля зрения на стороне плегии) – это следует учитывать при выборе места в классе для ученика с такой патологией. У многих пациентов с гемиплегической формой ДЦП кроме двигательных имеются значительные нарушения чувствительности (гемианестезия), и прогноз функциональной реабилитации пораженных конечностей часто в большей степени связан именно с гемианестезией, а не со спастикой. С нарушением чувствительности, а не с бездеятельностью связывают также отставание в росте руки на стороне плегии.
Умственная отсталость чаще выявляется у пациентов с пренатальным развитием гемиплегии, нежели с интра- и постнатальными гемиплегическими формами. В целом ее частота среди больных ДЦП составляет 65% случаев. Умственная отсталость выражена значительнее и особенно часто встречается при диплегической форме ДЦП у пациентов, родившихся в срок, по сравнению с родившимися преждевременно (это может указывать на роль перинатальных и постнатальных факторов развития ДЦП). Причина столь парадоксального различия окончательно, тем не менее, не установлена.
При дистонической форме ДЦП умственная отсталость встречается относительно редко – у 50% пациентов. Интеллект у больных с гиперкинетической формой ДЦП большей частью развивается без отклонений от нормы (в диапазоне от среднего до высокого). Однако из-за трудностей общения с пациентами в связи с выраженными дизартрией и гиперкинезами нелегко убедиться в отсутствии у них умственного недоразвития, равно как трудно установить нормальные показатели интеллектуального развития.
В группе больных с атактической формой ДЦП умственная отсталость встречается достаточно часто (и это сочетается обычно с высокой частотой эпилептических припадков). Больные ДЦП, начинающие ходить до достижения 2 лет, чаще имеют нормальный или пограничный интеллект. В целом чем более выражены двигательные нарушения, тем грубее нарушения интеллектуального развития (из этого правила есть исключения, но их немного).
Из других нарушений у пациентов ДЦП заслуживают упоминания слюнотечение, дефицит массы тела и дисфункция мочевого пузыря.
Слюнотечение может вызывать выраженное раздражение кожи, но еще большее значение имеет его негативный косметический эффект. Установлено, что причиной слюнотечения является не собственно гиперсаливация, а неэффективность глотания (поэтому антихолинергические препараты вызывают лишь неприятные побочные эффекты).
Дефицит массы тела, как показывают недавние исследования, в немалой степени связан с плохой моторикой оральной мускулатуры (псевдобульбарным параличом) – зондовое питание или гастростомия приводят к значительному увеличению массы тела, а в некоторых случаях – и роста.
Приблизительно у 40% пациентов ДЦП выявляется императивность позывов к уринации и недержание мочи, а у 6% – вялость мочеиспускания. Генез этих нарушений остается неясным, но наличие функционального энуреза как будто исключается (как и функционального энкопреза). При цистометрии, в частности, нейрогенный мочевой пузырь выявляется лишь в небольшом проценте случаев. Нередко встречающийся у пациентов ДЦП запор и вторичный энкопрез является результатом неспособности контролировать абдоминальные мышцы.
Свыше 90% младенцев с ДЦП доживают до взрослых лет. Полностью обездвиженные дети с выраженной умственной отсталостью, получающие питание через зонд, не доживают до 5-летнего возраста.
Диагноз и дифференциальный диагноз ДЦП базируется главным образом на наличии двигательного дефекта и задержки развития двигательной функции при непрогрессирующем течении заболевания, а также на данных неврологического и психиатрического обследования, указывающих на церебральный генез расстройства. Следует заметить, что в части случаев, особенно в течение 2-го года жизни, дети с ДЦП утрачивают некоторые двигательные функции, что обычно связано с развитием контрактур, избыточной прибавкой массы тела или отсутствием мотивации двигаться (особенно у едва передвигающихся детей).
Некоторые заболевания (атаксия-телеангиэктазия, синдром Леша-Нихана) могут первоначально также проявляться непрогрессирующими двигательными нарушениями. Прогредиентный характер этих заболеваний становится очевидным лишь после 3–4 лет.
Особое внимание следует уделять дифференциальной диагностике с болезнью Сегавы (ДОФА-чувствительной мышечной дистонией). Дети, страдающие этой патологией, развиваются нормально до 6 месяцев, а затем возникают флуктуирующие в течение дня дистонические нарушения, которые обычно полностью купируются небольшими дозами леводопы. У гипотоничных детей («синдром вялого ребенка») можно предполагать спинальную амиотрофию или врожденную миопатию. В таких случаях диагноз основывается на данных электромиографии и мышечной биопсии.
Лечение ДЦП только симптоматическое и базируется на многолетнем комплексе лечебной физкультуры, массажа и физиотерапии. Необходимо начинать его как можно раньше, а именно в период формирования статических и локомоторных функций, когда спастические явления выражены еще не резко, отсутствуют стереотипные патологические позы, деформации, контрактуры.
Для снижения мышечного тонуса используют реланиум, баклофен, иногда – рассечение задних корешков спинного мозга, а также методы, препятствующие развитию контрактур и деформаций в конечностях (парафиновые и озокеритовые аппликации, гипсовые повязки, лонгеты, туторы, валики, воротники). В ряде случаев проводят операции на сухожилиях с целью их удлинения. При спастических и дистонических формах ДЦП положительный эффект может быть достигнут при использовании ботокса (токсина ботулизма), вводимого в мышцы, с которыми связаны наиболее выраженные функциональные нарушения. При наличии медленных гиперкинезов и дистонии иногда удается получить положительный эффект от назначения циклодола, а также препаратов леводопы в небольших дозах. Разрабатываются и применяются методы соматосенсорной стимуляции, в частности ношение космического нагрузочного костюма «Пингвин» или его модификации «Адели» – это способствует исправлению положения центра тяжести тела пациента и нормализации позы стояния. Предполагается, что такое лечение способствует перестройке нервных связей в полушариях мозга и изменению межполушарных взаимоотношений.
Рекомендуется, кроме того, назначение препаратов, улучшающих метаболизм и микроциркуляцию в нервной ткани (глутаминовая кислота, витамины группы B, ноотропы, например энцефабол, церебролизин, глицин, гаммалон, ацефен и др.). Следует иметь в виду, что у части пациентов, получающих ноотропы, появляются при этом раздражительность, гиперактивность, нарушения сна, а также диспептические расстройства. Назначаются также противосудорожные препараты, антиагреганты, седативные средства. Речевые нарушения требуют занятий с логопедом. Важную роль играют меры социальной реабилитации: развитие навыков самообслуживания, общения, труда, самоконтроля, школьное обучение, адекватная профессиональная ориентация. Очень важны семейная психотерапия, формирование у родителей мотивации к организации необходимых занятий с пациентами, осведомленности относительно проявлений ДЦП, динамики, прогноза, терапии, необходима также , так как рождение больного ребенка часто является поводом для взаимных обвинений и причиной развода родителей.
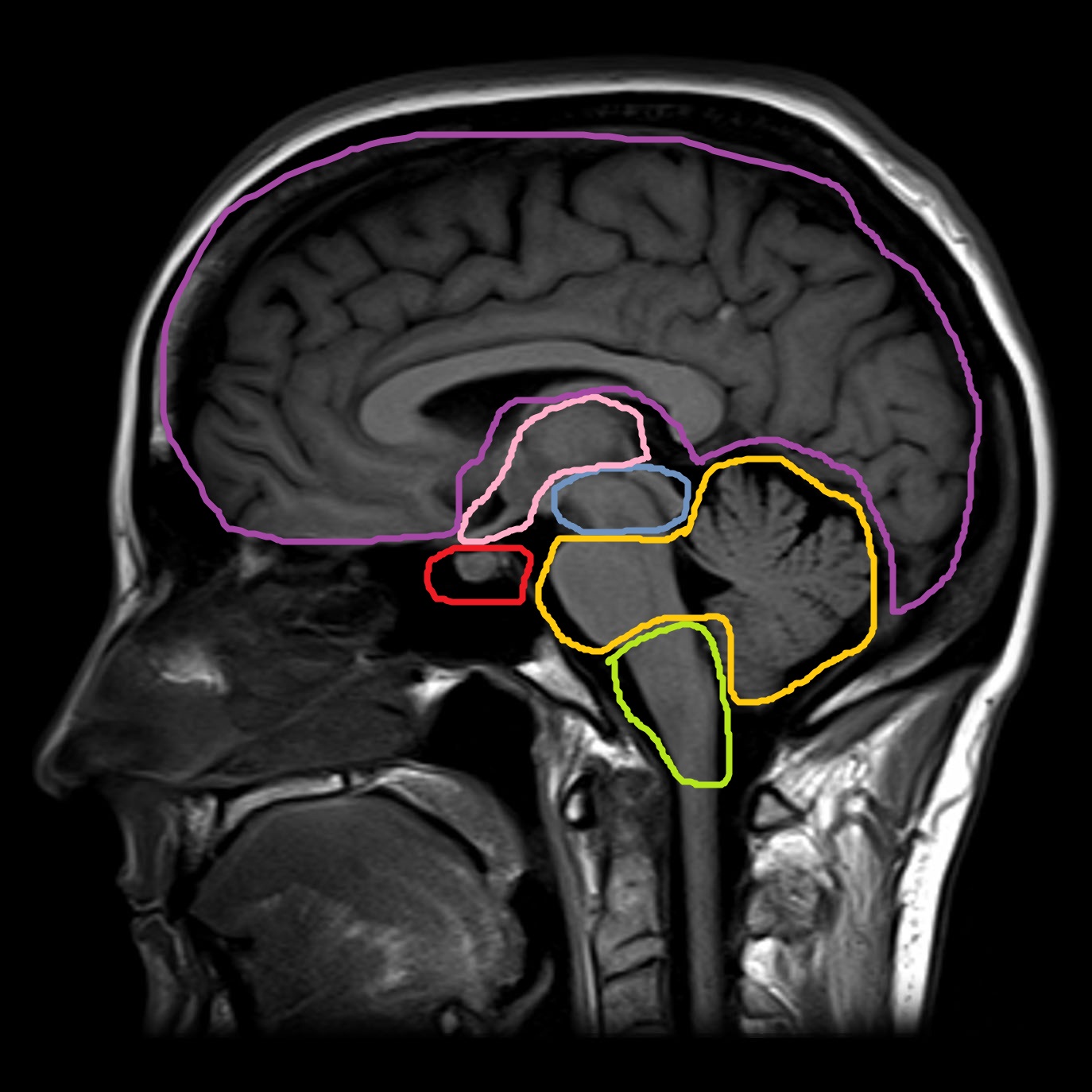
Основные морфологические отделы мозга
- передний (конечный) мозг состоит из двух больших полушарий.
- промежуточный мозг состоит из таламуса, эпиталамуса, гипоталамуса, гипофиза, который не включают в промежуточный мозг, а выделяют в отдельную железу.
- средний мозг состоит из ножек мозга и крыши четверохолмия. Верхние холмы крыши четверохолмия является подкорковым зрительным центром, а нижние холмы являются подкорковым центром слуха.
- задний мозг состоит варолиев мост и мозжечок.
- продолговатый мозг. Местом перехода продолговатого мозга в спиной мозг является большое затылочное отверстие.
Средний, задний и продолговатый мозг объединяют в ствол мозга.

Внутренняя структура больших полушарий.
- Серое вещество
- Белое вещество
Серое вещество состоит из коры, которое полностью покрывает большие полушарии головного мозга. Белое вещество расположено под серым веществом головного мозга. Однако в белом веществе также присутствуют участки с серым веществом - скопления нервных клеток . Их называют ядрами (nuclei). В норме существует четкая граница между белым и серым веществом. Дифференциация белого и серого вещества возможна на КТ, но лучше дифференцируется на МРТ.

Кортикальная дисплазия
При кортикальной дисплазии границы между белым и серым веществом стираются. В таком случае дополнительно следует использовать последовательность Т1 инверсия восстановления. На данных изображениях границы будут заметны, за исключением участков кортикальной дисплазии.

Инфаркт
При цитотоксическом отеке, развивающейся в первые минуты инфаркта головного мозга, также теряется дифференцировка между белым и серым веществом, что является ранним КТ признаком инфаркта головного мозга.







Большие полушария головного мозга
Полушария головного мозга разделяются между собой большим серповидным отростком. В каждом полушарии выделяют 4 доли:
- лобная доля.
- теменная доля
- затылочная доля
Лобная доля отделяется от теменной при помощи центральной или раландовой борозды, которая отлично визуализируется, как на аксиальных, так и на сагиттальных срезах.
Лобная доля отделяется от височной доли при помощи латеральной борозды, которая отлично визуализируется, как на сагиттальных и аксиальных, так и на фронтальных срезах.
Теменная доля отделяется от затылочной доли при помощи одноименной теменно-затылочной борозды. Данная линия еще разделяет каротидный и базиллярный бассейн.
Некоторые авторы в отдельную борозду выделяют островок, который является большим участком коры, покрывающий островок сверху и латерально, образует крышечку (лат. pars opercularis) и формируются из части прилегающих лобной, височной и теменной долей.
Границы долей


Границы долей
Границы лобных и теменных долей.
Омега - ?
Центральная борозда
Симптом усов – постцентральная извилина.
Поясная извилина – постцентральная извилина.
Для правильного определения границы лобных и теменных долей сначала находим центральную борозду. В данную борозду вписывается символ Омега – ? на аксиальных срезах.
Также помогают симптом усов, расположенных перпендикулярно срединной линии и изображение, которых соответствуют постцентральной борозде. Кпереди от постцентральной извилины расположена соответственно центральная борозда.
Поясная борозда.
На сагиттальных срезах нужно найти мозолистое тело над ним расположена поясная борозда, которая кзади и кверху продолжается в постцентральную борозду, от которой кпереди расположена центральная или роландова борозда.

Лобная доля
Лобная доля имеет большие размеры и одна из главных извилин является прецентральная извилина, являющаяся корковым центром движения. В лобной доле также отмечают верхнюю, среднюю и нижнюю извилины. Перечисленные извилины идут сверху вниз и параллельно друг другу.
На нижней поверхности лобной доле прямые и глазничные извилины, между которыми располагаются обонятельные тракты и луковицы. Данные области повреждаются при травмах.

Травматическое повреждение лобной доли
У данного пациента мы отмечаем симметричные повреждения базальных отделах обеих лобных долей, что соответствуют посттравматическим изменениям.

Зона Брока
Также важной зоной является зона Брока, которая расположена в дистальных отделах нижней лобной извилины. Ее локализация важна при планировании нейрохирургического вмешательствах. Данную зону легко найти, вспоминая о значке Макдональдс.

Инфаркт с вовлечением в патологический процесс зона Брока
У данного пациента острый инфаркт, обусловленный окклюзией передней ветви М2 левой СМА. Повреждения лобной доли с вовлечением в патологический процесс зоны Брока.

Теменная доля
Позади центральной борозды расположена постцентральная извилина, служащая корковым анализатором общей и проприоцептивной чувствительности.
Кзади расположены верхние и нижние теменные дольки.
В верхней теменной дольке располагается ядро кожного анализатора, ответственного за стереогнозию – способность узнавать предметы наощупь.
В нижней теменной дольке располагается двигательный анализатор, ответственный за апраксию – целенаправленные и произвольные движения.
Стереогнозия - способность узнавать предметы наощупь.
Апраксия - нарушение произвольных действий.

Атрофия предклинья
Атрофия предклинья является ранним симптомом болезни Альцгеймера еще до атрофии коры височных долей и гиппокампа.
Предклинье (Precuneus) участок теменной доли на внутренней поверхности обоих полушарий большого мозга, расположенный над мозолистым телом и впереди него.

Височная доля
В височной доле выделяют
Верхнюю височную извилину
Среднюю височную извилину
Нижнюю височную извилину. Данные три извилины параллельны друг другу и располагаются в горизонтальной плоскости.
Извилины Гешля, расположены на поверхности верхней височной извилины. Являются корковым центром слуха.
Парагиппокампальную извилину располагается на нижней поверхности височных долей в медиальных отделах. Крючок вместе с гиппокампом ответствены за обоняние. При повреждении гиппокампа нарушается память в первую очередь.
Зону Вернике. Зона Вернике расположена в дистальных отделах верхней височной извилины. Является сенсорной речевой зоной.

Затылочная доля
В затылочных долях определяются непостоянные борозды и извилины, но самая постоянная является шпорная борозда, расположенная на медиальной поверхности затылочной доли. Вокруг шпорной борозды расположены 17, 18 и 19 поля Бродмана, которые являются корковым центром зрения.

Окклюзия ЗМА
У данного пациента клинически отмечается нарушение зрения, обусловленные повреждением затылочной доли причиной, которой явился инфаркт (окклюзия ЗМА).
Подкорковое серое вещество







Подкорковое серое вещество
К подкорковому серому веществу относится:
- таламус
- базальные ядра
- хвостатое ядро
- лентикулярное ядро, в котором выделяют скорлупу и бледный шар.
- скорлупа
Внутренняя капсула состоит из переднего бедра, колено и заднее бедро.
Как найти заднее бедро?
Между таламусом и лентикулярным ядром находим гиперинтенсивный очаг, являющийся пирамидным трактом. От этого гиперинтенсивного очага проводим линию к колену, что и будет проекцией заднего бедра внутренней капсулы.
NB – не путать заднее колено с бледным шаром.
При классификации внутримозговых кровоизлияний в подкорковое серое вещество в зависимости от расположения по отношению к внутренней капсулы кровоизлияния делят на:
- латеральные
- медиальные
- смешанные
БЕЛОЕ ВЕЩЕСТВО
Комиссуральные волокна, с помощью которых полушария соединяются между собой.
Мозолистое тело (самая большая комиссура)
Передняя комиссура
Задняя комиссура (спайка свода)

Передняя комиссура
Передняя комиссура располагается под клювом мозолистого тела позади концевой пластинки и соединяет некоторые части обонятельного мозга: гиппокампальные извилины, левые и правые крючки височных долей.
Задняя комиссура
Задняя комиссура относится к эпиталамусу, находится у корня эпифиза и соединяет соответствующие части среднего и промежуточного мозга.
Практическое значение:
Для оценки мозолистого тела используется бикомиссуральная линия в сагиттальной плоскости. Бикомиссуральная линия проводится через верхний край передний коммисуры и нижней край задней комиссуры.

Мозолистое тело
Мозолистое тело состоит:
Ствол или тело (передний и задний отдел)
Каждый отдел соединяет гомолатеральный отдел головного мозга.
Формирование мозолистого тела.
Мозолистое тело развивается в особом порядке:
От колена, затем тела, валик и в конце развивается клюв.
Миелинизация мозолистого тела идет от задних отделов к передним отделам.
Данные знания помогают сузить дифференциальный диагноз при патологиях мозолистого тела.

Дисгенезия и атрофия мозолистого тела
При дисгенезии мозолистого тела хорошо сформировано колено и передние отделы мозолистого тела, но отсутствует валик и клюв. Данная патология является врожденной. Патология представлена слева.
При атрофии мозолистого тела хорошо сформированы задние отделы мозолистого тела (задний отдел тела и валик), но при этом уменьшены в размерах клюв, колено и передний отдел тела. Данные изменения являются приобретенными.
Многие заболевания поражают мозолистое тело, поэтому наличие очагов не являются патогномоничным для определенного заболевания.

Болезнь Маркиафавы-Биньями
Болезнь Маркиафавы-Биньями (центральная дегенерация мозолистого тела, Маркиафавы синдром, экстрапонтинный миелинолиз).
Встречается у лиц злоупотребляющих алкоголем. У данных лиц на МРТ выявляется поражение валика и задних отделов ствола (тела) мозолистого тела.
На хронических стадиях болезни Маркиафавы-Биньями визуализируется мозолистое тело в виде сэндвича, при котором сохраняется верхних и нижних слоев мозолистого тела, но с некрозом средних слоев.
Белое вещество
Белое вещество:
- перивентрикулярное
- глубокие отделы (семиовальные центры)
- U-волокна
Перивентрикулярное белое вещество расположено в непосредственной близости от боковых желудочков головного мозга.
U-волокна соединяют кору близлежащих извилин или субкортикальное белое вещество.
Глубокие отделы белого вещества расположенные между перивентрикулярным и субкортикальным белым веществом.
Очаги в белом веществе:
Очаги в белом веществе классифицируются в соответствии с локализацией:
- перивентрикулярные
- юкстакортикальные
- субкортикальные
- очаги в глубоком белом веществе

Перивентрикулярные очаги
перивентрикулярные (единичные или множественные, мелкие или крупные, сливающиеся между собой)
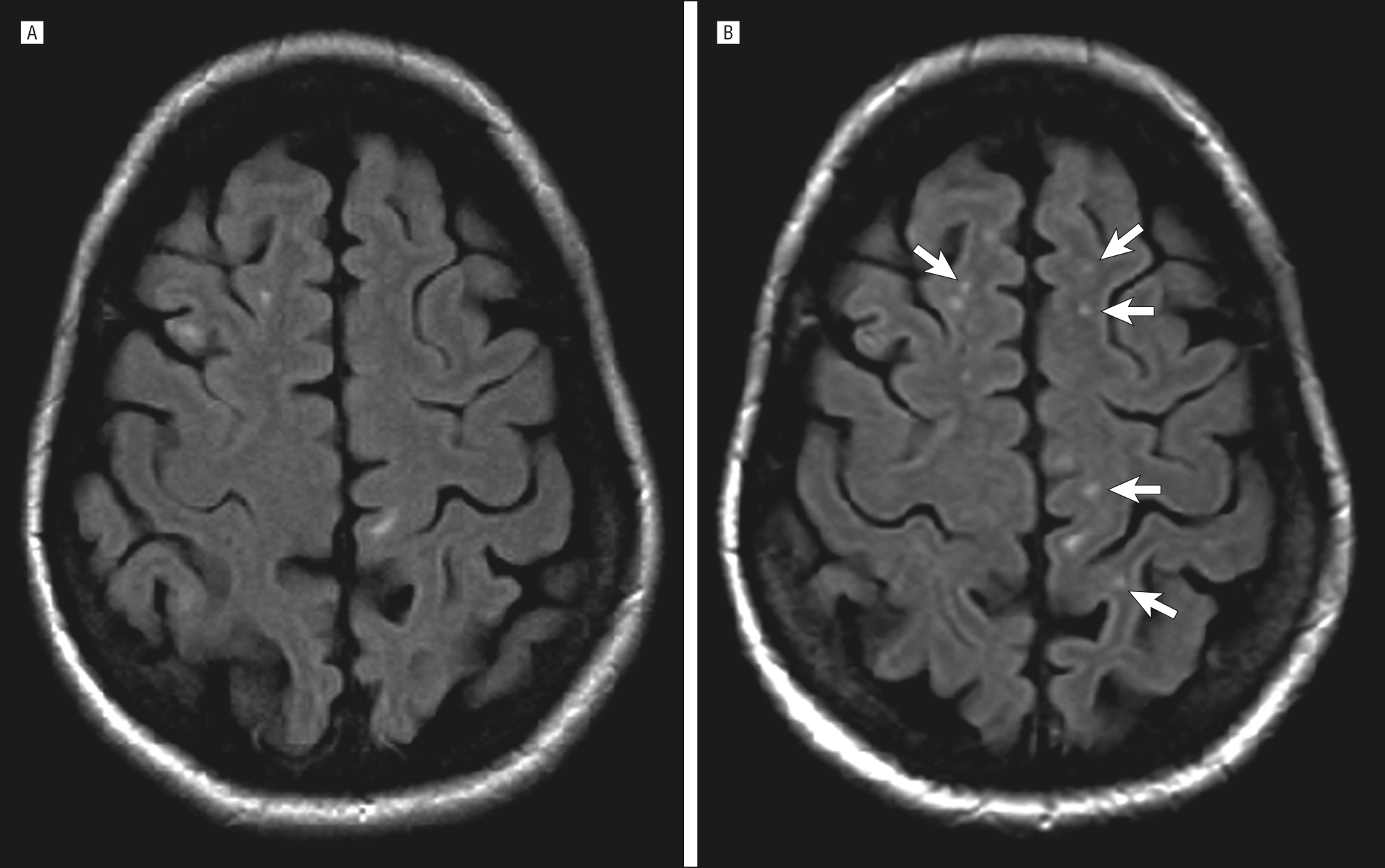
Юкстакортикальные очаги
juxta – около. Данные очаги локализуются в u-волокнах и непосредственно прилежат к серому веществу, то есть между очагом и серым веществом отсутствует прослойка белого вещества.
По форме данные очаги бывают разные, как повторять форму u-волокон, также могут округлой и неправильной формы. Данная локализация патогномонична для РС.

Субкортикальные очаги
Субкортикальные очаги – это очаги, которые локализуются вблизи коры головного мозга, но при этом между очагом и корой есть прослойка белого вещества.

Очаги в глубоком белом веществе.
Данные очаги встречаются при различных заболеваниях головного мозга.




ЖЕЛУДОЧКИ ГОЛОВНОГО МОЗГА
Боковые желудочки состоят из:
- передних (лобных) рогов
- задних (затылочных) рогов
- нижних (височных) рогов
Боковые желудочки соединяются с третьем желудочком с помощью парных отверстий Монро.
Третий желудочек имеет неправильную форму за счет наличия карманов. Отверстие третьего желудочка соответствует межталамической спайки.
Третий желудочек при помощи сильвиевого водопровода соединяется с четвертым желудочком. Из четвертого желудочка ликвор поступает в базальные цистерны через парные отверстия Люшка и непарную апертуру Можанди.
При оценки желудочков стоит обращать внимание на рога желудочков так, как при дегенеративных заболеваниях таких, как болезнь Альцгеймера, атрофия гиппокампа сопровождается с расширением височных рогов. В режиме FLAIR повышается сигнал от задних (затылочных) рогов, что является нормой также, как и асимметрия рогов.
ТРЕТИЙ ЖЕЛУДОЧЕК.
Третий желудочек располагается на срединной линии между зрительными буграми. Соединяется с боковыми желудочками посредством монроевых отверстий, с четвёртым желудочком посредством водопровода мозга.
Карманы третьего желудочка:
- Супрахиазмальный
- Инфундибуллярный
- Супрапинеальный
- Пинеальный
В норме данные карманы имеют острые углы, но при увеличении давления карманы раскрываются.
Четвертый желудочек головного мозга.
Четвертый желудочек является полостью заднего мозга и при помощи парных отверстий Люшка и непарного отверстия Мажанди соединяется с базальными цистернами.

Сосудистые сплетения
Сосудистые сплетения, продуцирующие ликвор, расположены во всех желудочках головного мозга, поэтому кальцификацию сосудистого сплетения, которая чаще визуализируется в задних рогах боковых желудочков, можно увидеть и в третьем, и в четвертом желудочке.

Туберозный склероз.
Не стоит путать обызвествление сосудистых сплетений, являющейся нормой, с патологическими состояниями. Например, с обызвествлениями боковых желудочков – перивентрикулярными туберсами при туберозном склерозе.

Гетеротопия серого вещества
Важно помнить, что единственное серое вещество, граничащее с боковыми желудочками – это хвостатые ядра, которые имеют четкие ровные контуры. Дополнительные структуры серого вещества, деформирующие контур боковых желудочков, являются патологическими изменениями, характерные при гетеротопии серого вещества.

Варианты строения желудочков
- полость прозрачной перегородки, которая отмечается у большинства новорожденных (закрывается со временем) и выглядит в виде треугольной формы между телами переднего бокового желудочка. Данная полость никогда ни пересекает отверстие Монро.
- полость промежуточного паруса. Одну из стенок полости, которой образует крыша третьего желудочка.
- полость Верге – это протяженная полость между телами боковых желудочков.

Коллоидная киста
Следует отличать варианты строения от коллоидной кисты, которая будет отличаться от интенсивности сигнала от ликвора практически во всех импульсных последовательностях. После введения контрастного вещества коллоидные кисты контраст не накапливают, что соответствует доброкачественному процессу.

МРТ норма - срединный сагиттальный срез. ЦСЖ - цистерны.
A - ЦИСТЕРНА КОНЦЕВОЙ ПЛАСТИНКИ
B - ЦИСТЕРНА ХИАЗМЫ
C - Межножковая цистерна
D - Обводная цистерна
Е - Квадригеминальная цистерна
F - Мостомозжечковая цистерна
G - Мостомозжечковая цистерна Prepontine pontocerebellaris Цистерна моста (препонтинная)
H - ЛАТЕРАЛЬНАЯ ЦЕРЕБЕЛЛОМЕДУЛЛЯРНАЯ ЦИСТЕРНА
I - ЦИСТЕРНА МАГНА
Изображение представлено Dr. Coenraad J. Hattingh
ЦИСТЕРНЫ ГОЛОВНОГО МОЗГА
Из четвертого желудочка головного мозга ликвор поступает в базальные цистерны при помощи парных отверстий Люшка и непарного отверстия Мажанди.
Название цистерн, исходя из локализации:
В сагиттальной плоскости:
- Супраселлярная цистерна
- Премостовая цистерна, в которой проходит основная артерия.
- Четверохолмная цистерна
- Большая или базальная цистерна мозга
В аксиальной плоскости:
- Межножковая цистерна
- Обводная цистерна соединяет межножковую и четверохолмную цистерны. Также у обводной цистерны выделяют крылья: правое и левое.
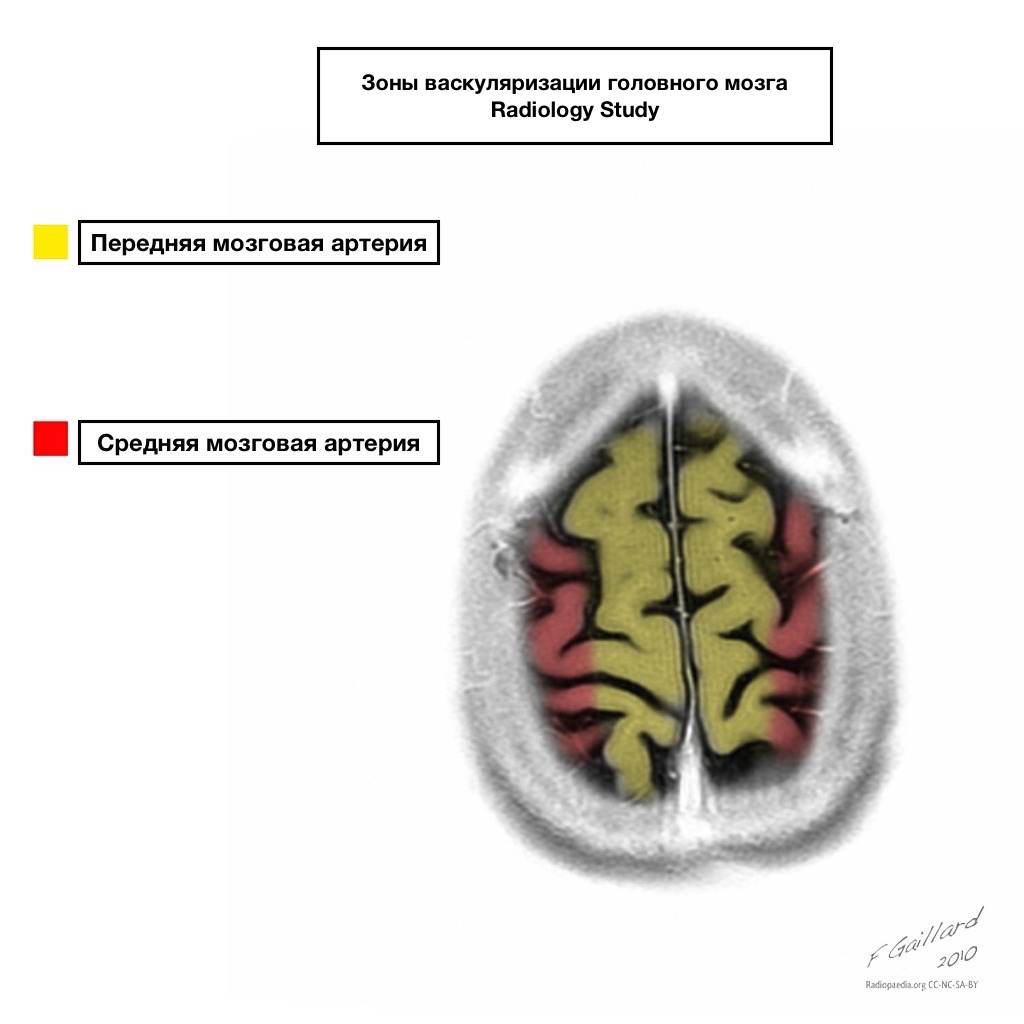









Бассейны кровоснабжения имеют четкие границы.

Зоны смежного кровоснабжения
Зоны смежного кровоснабжения на пересечении зон кровоснабжения:
Передней мозговой артерии
Средней мозговой артерии
Задней мозговой артерии.
Чаще всего инфаркта в данных зонах имеют гемодинамический характер, то есть отмечаются при падении АД.

Оболочки головного мозга
Головной мозг покрыт тремя оболочками.
- Мягкая оболочка плотно прилежит к головному мозгу, заходит во все щели и борозды, и в ней располагаются кровеносные сосуды. В определенных местах она проникает в желудочки мозга и образует сосудистые сплетения.
- Паутинная или арахноидальная оболочка ложится над бороздами и перекидывается с одной извилины на другую.
- Твердая оболочка изнутри выстилает полости черепа, плотно прилежит к ним и формирует венозные синусы и отростки, отделяющие отдельные структуры головного мозга друг от друга.
В норме оболочки головного мозга не визуализируются при МРТ, но после введения контраста твердая оболочка контрастируется.

Изменения мягких мозговых оболочек.
При лептоменингеальном карциноматозе на Т1 и Т2 безконтрастных изображениях отмечается повышение сигнала от мягких мозговых оболочек, а после введения контраста улучшает визуализацию.

Лептоменингит
Изменения мягких мозговых оболочек также нередко встречается при воспалительных изменения, например, при туберкулезном лептоменингите.

Изменение твердой мозговой оболочки
Изменение твердой мозговой оболочки встречается при интракраниальной гипотензии. При данной патологии визуализируется утолщенная твердая мозговая оболочка, интенсивно накапливающая контраст. Дополнительными критериями в постановке диагноза является увеличение в размерах гипофиза, пролабирование миндалин мозжечка в большое затылочное отверстие.

Изменение твердой мозговой оболочки также встречается при пахименингеальном карциноматозе, что проявляется утолщением твердой мозговой оболочки с интенсивным накоплением контрастного вещества и вазогенный отек, прилежащих отделов лобной доли.
![]()
Оболочечные пространства.
Оболочечные пространства – это пространства между оболочками головного мозга.
- Субарахноидальное пространство – это пространство между мягкой и арахноидальной оболочкой. В норме должна иметь интенсивность ликвора.
- Субдуральное пространство – это пространство между арахноидальной и твердой оболочкой.
- Эпидуральное пространство – это пространство между твердой оболочкой и костями черепа, которое в норме не визуализируется так, как твердая оболочка сращена с костями черепа.

Изменение субарахноидального пространства
Изменение субарахноидального пространства
Сужение. Данные изменения встречаются при объемном воздействие (опухоль, инфаркт).
Расширение. Данные изменения встречаются в посттравматический период, после инфаркта, либо при атрофии.



Оболочечные кровоизлияния
При оболочечных кровоизлияниях мы отлично можем выявить оболочки.
Виды оболочечных кровоизлияний:
Эпидуральное кровоизлияние. Обычно визуализируются в виде линзы и не распространяются за пределы швов, но могут пересекать синусы головного мозга, что является отличительной чертой от субдуральных кровоизлияний, которые никогда ни пересекают синусы головного мозга.
Субдуральное кровоизлияние. Наиболее частыми причинами является разрыв поверхностных вен в результате смещение мозга при травмах. Если в данном случае рвется и субарахноидальная оболочка, то в данном случае в субдуральное пространство попадает ликвор.
Субарахноидальное кровоизлияние. Выявляется повышение сигнала от ликвора в режиме FLAIR. Наиболее частыми причинами субарахноидального кровоизлияния является разрыв аневризмы так, как артерии, кровоснабжающие головной мозг, локализуются именно в субарахноидальном пространстве.

При патологических процессах в оболочках не используется термин доли, а вместо этого используется термин область. Например, у данного пациента менингиома лобной области.
Компот из вишни на зиму в банках (8 рецептов)
Компот из облепихи: вкусный защитник здоровья вашего ребенка Можно ли делать компот из облепихи
К чему снится салат из овощей: разбираемся в сновидении
Мифы о сотворении мира египетская мифология
Выручка в зарубежных странах